Prions:

Prions are responsible for the transmissible spongiform encephalopathies in a variety of mammals, including bovine spongiform encephalopathy (BSE, also known as "mad cow disease") in cattle. In humans, prions cause Creutzfeldt-Jakob Disease (CJD), variant Creutzfeldt-Jakob Disease (vCJD), Gerstmann–Sträussler–Scheinker syndrome, Fatal Familial Insomnia and kuru. All known prion diseases in mammals affect the structure of the brain or other neural tissue and all are currently untreatable and universally fatal.
Prions are not considered living organisms but are misfolded protein molecules which may propagate by transmitting a misfolded protein state. If a prion enters a healthy organism, it induces existing, properly folded proteins to convert into the disease-associated, misfolded prion form; the prion acts as a template to guide the misfolding of more proteins into prion form. These newly formed prions can then go on to convert more proteins themselves; this triggers a chain reaction that produces large amounts of the prion form. All known prions induce the formation of an amyloid fold, in which the protein polymerises into an aggregate consisting of tightly packed beta sheets. Amyloid aggregates are fibrils, growing at their ends, and replicating when breakage causes two growing ends to become four growing ends. The incubation period of prion diseases is determined by the exponential growth rate associated with prion replication, which is a balance between the linear growth and the breakage of aggregates.
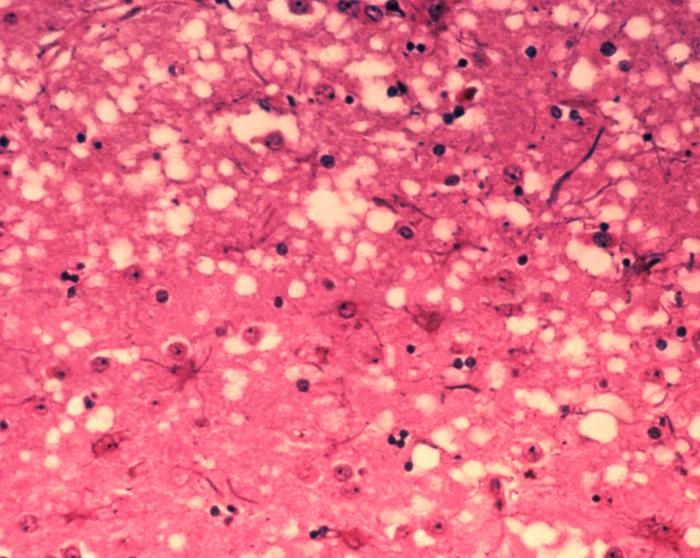
Prions cause neurodegenerative disease by aggregating extracellularly within the central nervous system to form plaques known as amyloid, which disrupt the normal tissue structure. This disruption is characterized by "holes" in the tissue with resultant spongy architecture due to the vacuole formation in the neurons. Other histological changes include astrogliosis and the absence of an inflammatory reaction. While the incubation period for prion diseases is relatively long (5 to 20 years), once symptoms appear the disease progresses rapidly, leading to brain damage and death.Neurodegenerative symptoms can include convulsions, dementia, ataxia (balance and coordination dysfunction), and behavioural or personality changes.
Transmission:
It has been recognized that prion diseases can arise in three different ways: acquired, familial, or sporadic. It is often assumed that the diseased form directly interacts with the normal form to make it rearrange its structure. One idea, the "Protein X" hypothesis, is that an as-yet unidentified cellular protein (Protein X) enables the conversion of PrPC to PrPSc by bringing a molecule of each of the two together into a complex.
Current research suggests that the primary method of infection in animals is through ingestion. It is thought that prions may be deposited in the environment through the remains of dead animals and via urine, saliva, and other body fluids. They may then linger in the soil by binding to clay and other minerals.
Sterilization:
Infectious particles possessing nucleic acid are dependent upon it to direct their continued replication. Prions, however, are infectious by their effect on normal versions of the protein. Sterilizing prions, therefore, requires the denaturation of the protein to a state in which the molecule is no longer able to induce the abnormal folding of normal proteins. In general, prions are quite resistant to proteases, heat, radiation, and formalin treatments, although their infectivity can be reduced by such treatments. Effective prion decontamination relies upon protein hydrolysis or reduction or destruction of protein tertiary structure. Examples include bleach, caustic soda, and strongly acidic detergents such as LpH. 134 °C (274 °F) for 18 minutes in a pressurized steam autoclave may not be enough to deactivate the agent of disease. Ozone sterilization is currently being studied as a potential method for prion denaturation and deactivation. Renaturation of a completely denatured prion to infectious status has not yet been achieved; however, partially denatured prions can be renatured to an infective status under certain artificial conditions
The World Health Organization recommends any of the following three procedures for the sterilization of all heat-resistant surgical instruments to ensure that they are not contaminated with prions:
Immerse in a pan containing 1N NaOH and heat in a gravity-displacement autoclave at 121 °C for 30 minutes; clean; rinse in water; and then perform routine sterilization processes.
Immerse in 1N NaClO (sodium hypochlorite) (20,000 parts per million available chlorine) for 1 hour; transfer instruments to water; heat in a gravity-displacement autoclave at 121 °C for 1 hour; clean; and then perform routine sterilization processes.
Immerse in 1N NaOH or sodium hypochlorite (20,000 parts per million available chlorine) for 1 hour; remove and rinse in water, then transfer to an open pan and heat in a gravity-displacement (121 °C) or in a porous-load (134 °C) autoclave for 1 hour; clean; and then perform routine sterilization processes.
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου